




A BRAVE Sheldon youngster with a life-threatening rare, and at one-time fatal, genetic condition is now thriving after a ‘world first’ new enzyme treatment.
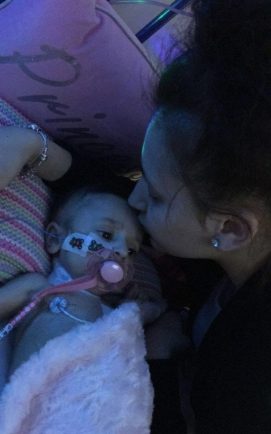
Inaaya Shabbir has Wolman’s Disease, a condition that unless treated rapidly, causes multi-organ damage, leading to death in early infancy.
It occurs in around one in 350,000 births and is a type of lysosomal acid lipase deficiency, which causes a build-up of fat in cells in the liver, heart, blood vessels and digestive system.
However, due to Sebelipase alfa, an recombinant enzyme replacement therapy at Birmingham Children’s Hospital, which works by replacing the deficient enzyme, seven-year-old Inaaya has gone well beyond the years predicted for her when she was diagnosed.
No child with this condition had previously survived beyond six months of age before this new treatment was developed. Now, children can be expected to have normal development and their life expectancy is likely to be the comparable to that of the general population.
A commercial deal struck by NHS England in November recommended Sebelipase alfa for use on the NHS in final draft guidance by the National Institute for Health and Care Excellence (NICE).
Inaaya’s mum, Amber Khan, said: “When Inaaya was diagnosed it felt like my whole world was breaking down.
“When I had her, I’d already had a son who was healthy and there was less than two years between them, so I didn’t know what was going wrong, but after a week I noticed her belly was big, but her arms and legs were small.
“She came back and told me she had Wolman’s and to not look at it online. I did and told my mum, all it talked about was death. I was scared. The team took Inaaya for a blood transfusion and I was told I’d get a call from a consultant at the Children’s Hospital.”
Little did Amber know but she was in the right place.
Birmingham Children’s Hospital was one of the centres pioneering the treatment, alongside the Royal Manchester Children’s Hospital, and one of the few in the world that had seen multiple patients with this rare condition.
Dr Suresh Vijay, Consultant of Clinical Paediatric Inherited Metabolic Disorders at Birmingham Women’s and Children’s NHS foundation Trust, led the study.
He said: “Until around 2014 there was no treatment at all for Wolman’s and all affected babies would show symptoms in their first few weeks to months of life. Sadly, none would survive longer than three or four months old.
“Now we have children who are living around Inaaya’s age and older, so it has been a huge development.
Amber added: “Inaaya is so amazing, nothing can phase her. She is so selfless, she can’t eat chocolate and sweets, but she would happily give sweets out to everyone in her class, even knowing she can’t have it. I’m so lucky to have her.”










